科学家开发出一款分子模拟新工具
更多精彩请关注公众号:分子动力学
Dynamo分子建模和模拟库的开发始于20世纪90年代初。这项工作的目标是实现一个可以用混合量子化学(QC)和分子力学(MM)方法进行分子模拟的程序,因为在那时,软件的选择十分有限。
Dynamo的第一个版本,即现在的fDynamo,是用Fortran90/95语言编写的,并于20世纪90年代末发布。
fDynamo实际上不是一个独立的程序,而是一个Fortran模块库,用户可以使用它来为他们想要执行的每个特定的模拟编写自己的程序。
在考虑了多种语言之后,Python版本的Dynamo在21世纪初开始开发,并于2007年发布了pDynamo的第一个版本,随后几年又发布了几个增量维护版本。

2022年11月30日,法国约瑟夫傅立叶大学Martin J Field研究者发表成果,介绍了pDynamo库自2007年以来的第一次重大修订,称为pDynamo3,这是一款基于Python的分子建模与模拟程序。
pDynamo及其前身fDynamo的设计目的都是为在原子水平上进行分子模拟提供易于使用和灵活的框架,特别是那些采用混合量子化学和分子力学的方法。
尽管pDynamo3的使用与pDynamo2十分相似,但它增加了重要的新功能,并进行了广泛的重组,这将使它更容易扩展新功能。
pDynamo3代码根据GNU发布网址为https://github.com/pdynamo/pDynamo3。
pDynamo3保留了pDynamo2的所有功能。它允许使用QC、MM和QC / MM势对原子体系进行大量的能量和力计算,包含广泛的模拟算法,如几何优化、分子动力学、正态分析和反应路径搜索,并拥有一套重要的分析算法。
模拟是通过编写Python脚本来执行的,这些脚本利用了pDynamo3库中可用方法的多样性,并在必要时与那些可以在广泛的第三方Python模块和包中找到的方法相结合。
作为一个非常简单的例子,考虑图1所示的pDynamo3脚本,使用三种不同的半经验QC方法计算水分子的能量和性质。

图中的脚本只做了相对有限的计算,但经验表明,各种甚至是概念上高度复杂的模拟和分析都可以在相当简洁的pDynamo3脚本中直接有效地进行,而不需要求助于更专业的模拟程序。
尽管pDynamo3表面上与pDynamo2相似,但pDynamo3经历了彻底的内部修改。其中最重要的变化有以下几点:
1、通过引入特定于有限数量任务的子包对pDynamo的包进行重组。图2给出了当前软件包及其子包的示意图,以及pDynamo3发行版中其他重要的非软件包目录的示意图。
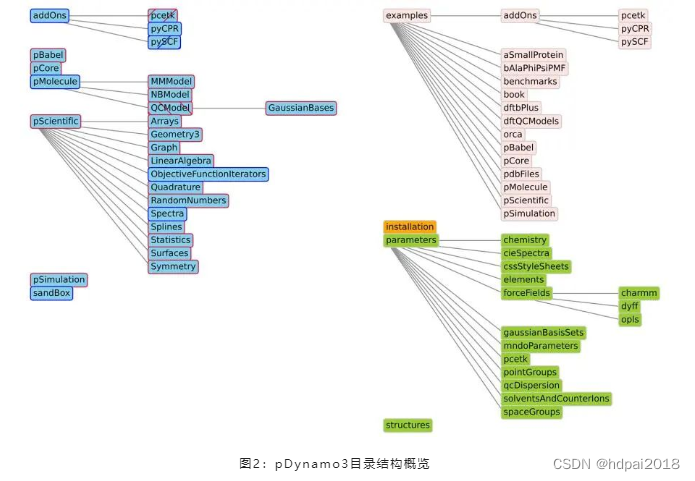
2、一个简化的安装程序,自动安装所有的包和子包,并在pDynamo的目录树中找到正确的结构。此外,安装脚本允许部分和全部安装,这对开发和测试特别有用。
3、将所有pDynamo的例子和测试重新组合到一个目录中,并改进了脚本,可以运行所有的例子和测试,或者只运行特定包或主题的子集。
Dynamo计划的最初目标之一是拥有一个平台来开发和应用执行QC / MM模拟的方法。尽管pDynamo3具有其他重要的功能,但它在这方面的能力特别强大。
尤其是主要分布中所有的MM、QC和非键合相互作用方法具有一致的界面,因此它们可以在QC / MM计算中结合并互换使用。
作为其中一些近似的例子,使用pDynamo3的态链反应路径方法进行了反应路径计算,使用了一个MM,三个QC和七个QC / MM能量模型,用于丁烷的反式构象转变和环己烷的椅式构象转变,所得能量曲线图如下图所示。
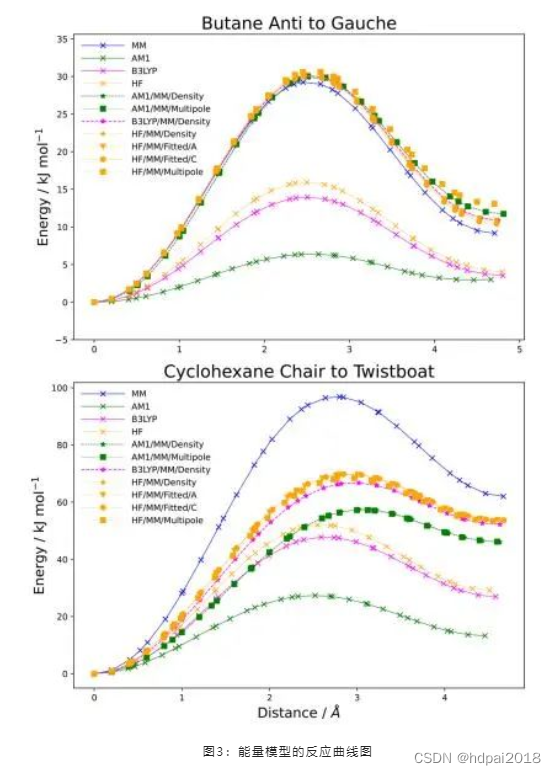
总而言之,该程序被设计为具有灵活和可扩展的结构,目的是确保它能跟上分子建模和模拟领域的最新发展,从而在未来继续成为一个有用的工具。
本文来自互联网用户投稿,文章观点仅代表作者本人,不代表本站立场,不承担相关法律责任。如若转载,请注明出处。 如若内容造成侵权/违法违规/事实不符,请点击【内容举报】进行投诉反馈!