生物芯片---ChAMP包学习笔记
文章目录
- 一、甲基化芯片简介
- 二、ChAMP包简介
- 三、具体处理流程
- (一)数据准备
- 1.使用ChAMPdata包中的数据
- 2.数据库下载,以GEO为例说明数据准备
- (二)数据读取与过滤`champ.load`
- (三)可视化查看质量控制情况`champ.QC`
- (四)数据标准化 `champ.norm`
- (五)批次效应
- 1.查看是否有批次效应`champ.SVD`
- 2.校正批次效应`champ.runCombat`
- (六)甲基化分析
- 1.champ.DMP
- 2.champ.DMR
- (七)找拷贝数变异CNA`champ.CNA`
- (八)GSEA分析`champ.GSEA`
一、甲基化芯片简介
一张芯片包括12个array,也就是一张芯片可以做12个sample,一台机子可以跑8张芯片,也就是一共96个sample,每个样本可以测到超过450,000个CpG位点的甲基化信息(大概人所有的1%,但是覆盖了多数CpG岛和启动子区),芯片本身包含一些控制探针可以做质控。
芯片注释信息:
#查看450k芯片注释信息
library(IlluminaHumanMethylation450kmanifest)
show(IlluminaHumanMethylation450kmanifest)#查看850k芯片信息使用另一个R包
library(IlluminaHumanMethylationEPICkmanifest)
minfi包中有特定的函数getManifest()可以查看指定对象对应芯片的注释信息
二、ChAMP包简介
ChAMP可以用来处理450k和EPIC的数据,包中整合了质量控制、数据标准化、识别DMR、识别CNA等函数,使甲基化数据的处理形成一个pipeline。
如果你的整个流程中参数都设置为默认值,那么你只需运行函数:champ.process(directory = testDir),参数directory为甲基化原始数据.IDAT所在文件目录。
当然,ChAMP中的每个函数也可以单独运行,自定义参数。
三、具体处理流程
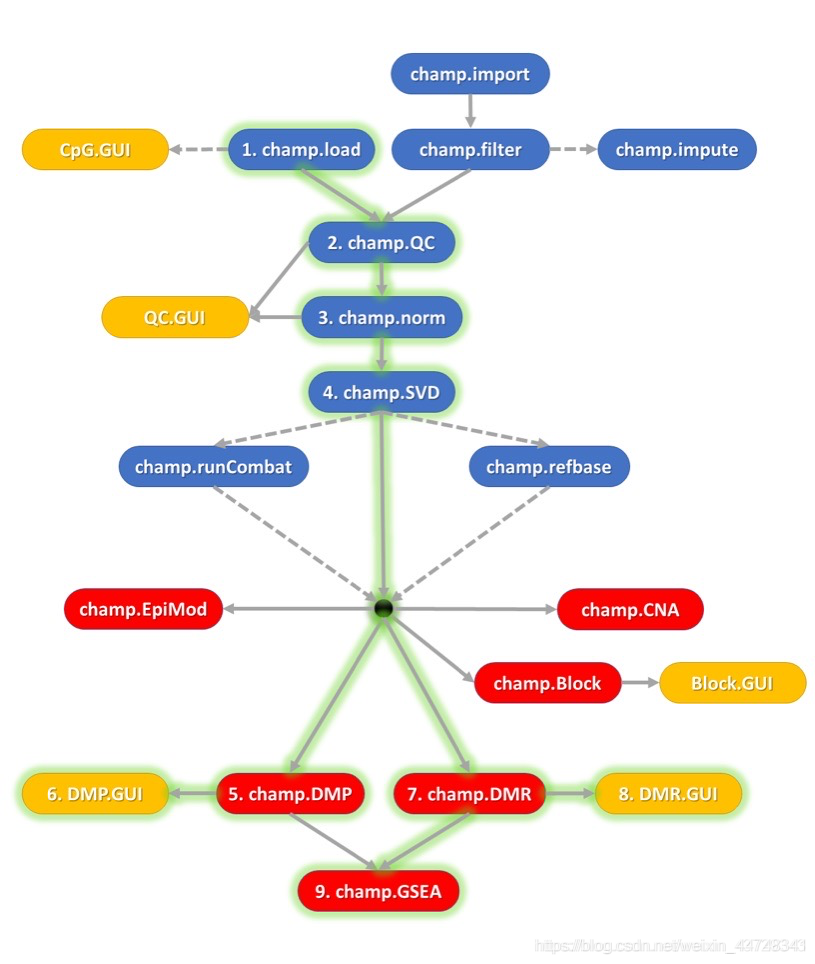
(一)数据准备
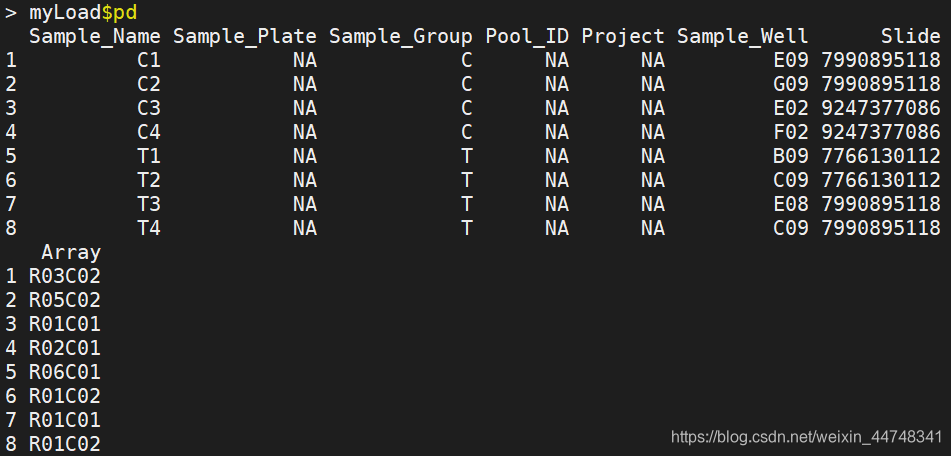
ChAMP包读取数据不仅要.idat原始文件,还需要一个pd(phenotype)文件,名为sample_sheet.csv(当然sample_sheet这个名字不重要,重要的是这是一个csv文件),具体信息如下:
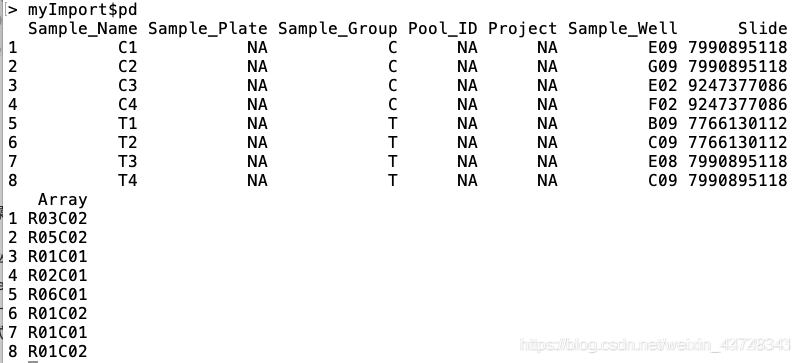
其中比较重要的属性列是Sample_Name、Sample_Group、Slide(Sentrix_ID)、Array(Sentrix_position)
Array(Sentrix_position):一张甲基化芯片上最多有12个样本,每个样本根据Sentrix_position标识;
Slide(Sentrix_ID):当样本个数大于12个时,必然需要另外一张芯片,对于每张芯片,使用Sentrix_ID标识;
minfi就是通过Sentrix_ID和Sentrix_position这两个字段来查找样本的原始数据
需要根据各样本信息自行创建一个sample_sheet.csv,如何构建?Sentrix_ID 和 Sentrix_Position来自IDAT文件名。
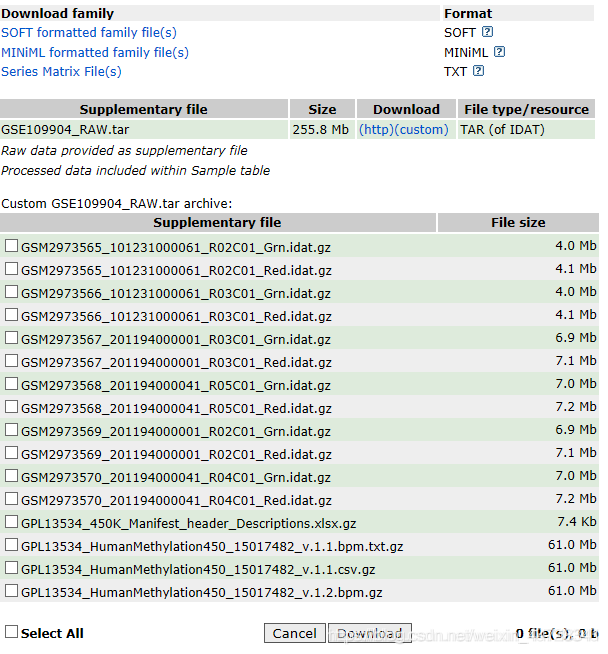
注意:GEO下载的数据命名是提交者自己命名,难以构建该csv文件。TCGA下载数据,文件名包含所需信息,可以构建该csv文件
1.使用ChAMPdata包中的数据
library(ChAMP)
testdir<-system.file("extdata",package="ChAMPdata")
去上述路径查看数据:
testDir[1]="/public/workspace/shaojf/anaconda3/lib/R/library/ChAMPdata/extdata"
包含8个样本,4个肿瘤样本,4个对照样本
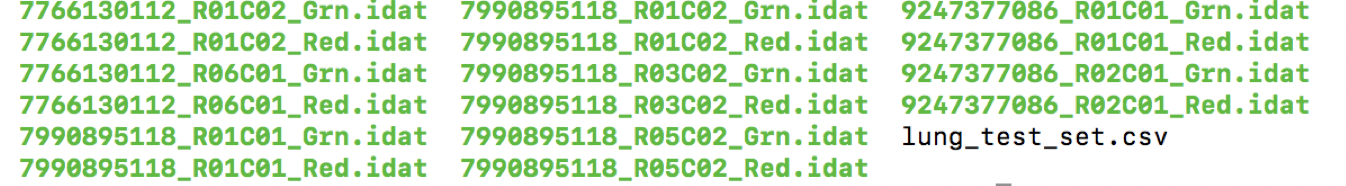
2.数据库下载,以GEO为例说明数据准备
(二)数据读取与过滤champ.load
myLoad <-champ.load(testDir,arraytype="450K")
#芯片类型,可设置为"EPIC"用于加载850K芯片的数据
champ.load()参数见champ.import()和champ.filter(),champ.load()利用minfi包来读入数据
champ.load()==champ.import()+champ.filter()
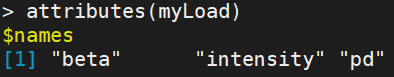
返回一个列表包含3个元素:“beta”,“intensity”,“pd”
beta是一个矩阵,列代表样本,行代表探针
pd记录样本信息,即自己编写的sample_sheet.csv文件,重要的属性列"Slide",“Array”
读入数据后有6种数据过滤:
① filterDetP、detPcut:默认过滤掉<0.01的探针,使用detPcut来修改阈值
②filterBeads、beadCutoff:探针与DNA序列杂交,beadcounts就是与探针结合的reads数,若结合得较少,则认为该种探针信号不可靠,实际处理中,默认如果这个探针在5%的样本中,beadcount<3这个探针就会被过滤掉,使用beadCutoff来修改阈值
③ filterNoCG:过滤非CpG位点的探针,对甲基化的C而言,有CpG\CHG\CHH等甲基化的C,若不想过滤这些探针可以设置filterNoCG=FALSE
④ filterSNPs:过滤SNP附近的CpG位点的探针,SNP位点附近的CpG位点不稳定,设filterSNPs=FALSE不进行过滤
⑤filterMultiHit:过滤非特异性的探针,Nordlund等研究人员使用bwa将探针序列与基因组进行比对,若唯一比对,说明探针特异性好,根据比对结果,去除比对到基因组上多个位置的探针,修改参数filterMultiHit=FALSE不进行过滤
⑥filterXY:过滤性染色体上的探针,性染色体上的差异CpG是不准确的,无法解释这个差异是否是实验造成,若性别相同时,可以不用过滤,修改参数filterXY=FALSE不进行过滤
(三)可视化查看质量控制情况champ.QC
champ.QC(beta = myLoad$beta,#---产生的β矩阵pheno=myLoad$pd$Sample_Group,#---样本分类信息mdsPlot=TRUE,densityPlot=TRUE,dendrogram=TRUE,PDFplot=TRUE,Rplot=TRUE,Feature.sel="None",#---绘制dendrogram时所使用的计算方法resultsDir="./CHAMP_QCimages/")
QC.GUI()
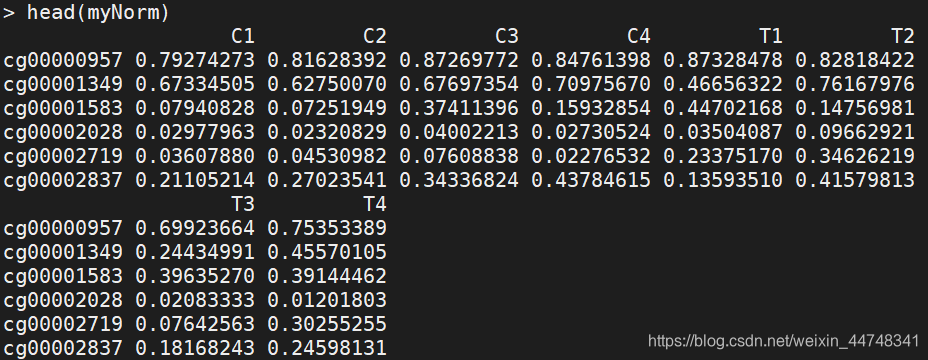
(四)数据标准化 champ.norm
对探针的矫正即消除两类探针因技术差异带来的误差
myNorm<-champ.norm(beta=myLoad$beta,rgSet=myLoad$rgSet,mset=myLoad$mset,resultsDir=".CHAMP_Normalization/",method="BMIQ",plotBMIQ=FALSE,arraytype="450K",cores=3)
(五)批次效应
1.查看是否有批次效应champ.SVD
champ.SVD(beta = myNorm,rgSet=NULL,pd=myLoad$pd,RGEffect=FALSE,#---If Green and Red color control probes would be calculatedPDFplot=TRUE,Rplot=TRUE,resultsDir="./CHAMP_SVDimages/")
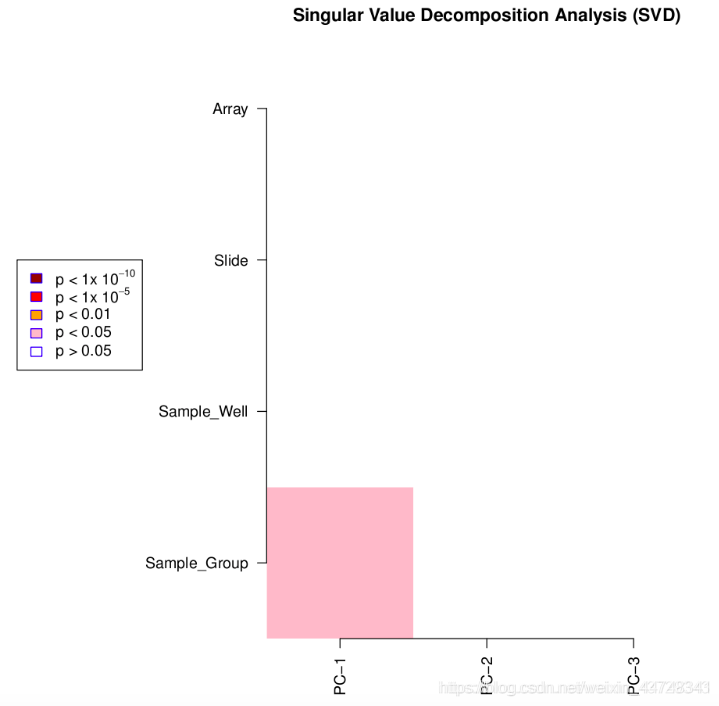
产生2个图像:Singular Value Decomposition Analysis (SVD);Scree Plot
①Singular Value Decomposition Analysis (SVD)
颜色越深,p-value越小,变异越显著,表明变异成分越大
从图像可以看出变异主要来自样本间(control组和tumor组),即我们关注的生物学因素,但变异显著性不大,经过过滤后得到的数据比较clear,适合后续分析。
如果发现在array或slide等有较显著的p-value,则需要检查实验设计以及使用其他normalization methods来进行后续的批次效应矫正
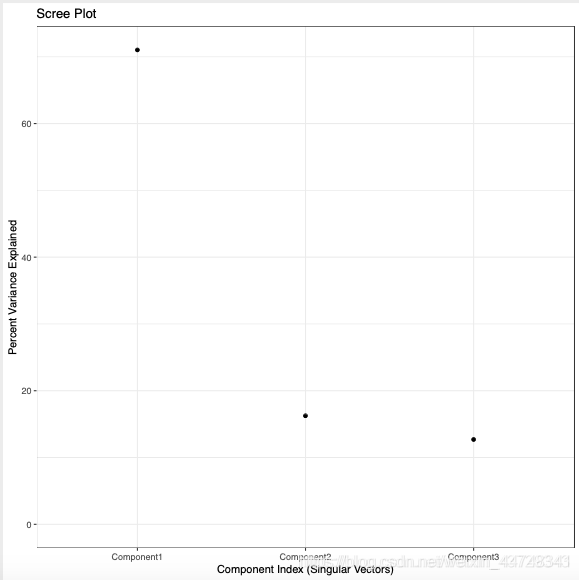
②scree plot:
各主成分可解释的变异占总变异的百分比
2.校正批次效应champ.runCombat
mycombat <- champ.runCombat(beta=myNorm,pd=myLoad$pd,variablename="Sample_Group",#要进行矫正的协变量,参考sva包的ComBat函数batchname=c("Slide"),#批次,来自pd文件logitTrans=TRUE)#对beta是否进行logit转化,利用原始beta进行矫正时设为T,利用M值进行计算时设为F
ComBat如果直接用beta值进行矫正,输出值可能不在0-1之间,所以在计算之前会进行一个logit转化成M-value;
若用M值进行矫正则没必要进行转化,因此设置logitTrans=F
(六)甲基化分析
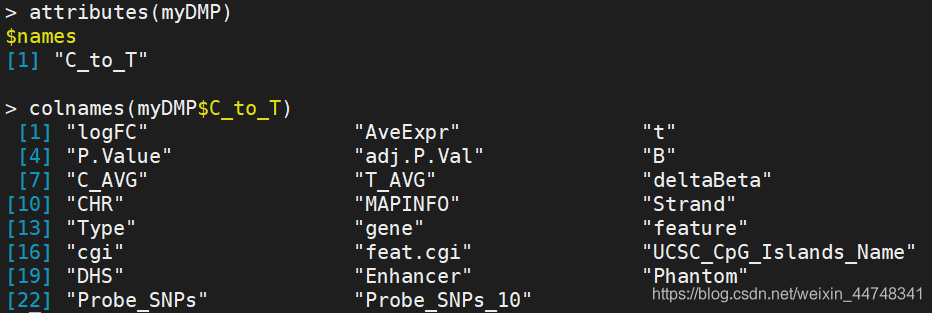
1.champ.DMP
myDMP <- champ.DMP(beta = myNorm,pheno = myLoad$pd$Sample_Group,compare.group = NULL,#当group>2时设定该参数为一个列表,其中每个元素为两个Sample_Group,进而指定要进行比较的group,否则每两个group之间都会进行比较adjPVal = 0.05,#设定的p-value检验水平adjust.method = "BH",#p-value矫正,使用的method参考函数p.adjust()arraytype = "450K")
#直接通过基于类别group的探针比较计算得到DMP# 可视化查看分布
DMP.GUI(DMP=myDMP[[1]],beta=myNorm,pheno=myLoad$pd$Sample_Group)
#使用GUI需要shiny包
2.champ.DMR
myDMR<-champ.DMR(beta=myNorm,pheno=myLoad$pd$Sample_Group,compare.group=NULL,arraytype="450K",method = "Bumphunter",minProbes=7,#---DMR包含很多CpG位点,每个位点都对应一个探针,minProbes设置每个DMR包含的最小探针数adjPvalDmr=0.05,#---DMR的p-value是用Stouffler's method计算得到,再进行矫正后得到adjPvalDmrcores=3,## following parameters are specifically for Bumphunter method.maxGap=300,cutoff=NULL,pickCutoff=TRUE,smooth=TRUE,smoothFunction=loessByCluster,useWeights=FALSE,permutations=NULL,B=250,nullMethod="bootstrap",## following parameters are specifically for probe ProbeLasso method.meanLassoRadius=375,minDmrSep=1000,minDmrSize=50,adjPvalProbe=0.05,Rplot=T,PDFplot=T,resultsDir="./CHAMP_ProbeLasso/",## following parameters are specifically for DMRcate method.rmSNPCH=T,fdr=0.05,dist=2,mafcut=0.05,lambda=1000,C=2)# 可视化查看分布
DMR.GUI(DMR=myDMR)
(七)找拷贝数变异CNAchamp.CNA
(八)GSEA分析champ.GSEA
本文来自互联网用户投稿,文章观点仅代表作者本人,不代表本站立场,不承担相关法律责任。如若转载,请注明出处。 如若内容造成侵权/违法违规/事实不符,请点击【内容举报】进行投诉反馈!