鬼臼毒素通过减少DNA损伤和减少PI3K抑制作用可作为抗肿瘤候选药物
摘要:替尼泊苷是FDA批准的药物,被用于癌症治疗,但在长期临床试验中却伴随着强烈的副作用。本研究主要是通过分析替尼泊苷与DNA拓扑异构酶Ⅱ的结合模式,对替尼泊苷的分子结构进行修饰,从而发现潜在的低毒候选药物。生物实验表明,化合物15和16在人正常细胞上的IC50为120.4-125.1μMμM,与替尼泊苷相比(IC50 = 11.5-22.3 μM),具有较低的毒性。另外,体内研究表明,化合物15和16在HepG2细胞异种移植模型中能显着抑制肿瘤生长,而没有表现出明显的毒性(LD50值为208.45和167.52 mg/kg),低于替尼泊苷的毒性(LD50 = 46.12 mg/kg)。化合物15和16能引起轻微的γH2AX磷酸化,具有较低的DNA毒性和较低的PI3K/AKT抑制,所以化合物15和16可能是潜在的低毒性抗肿瘤药物。
分子对接(Molecular docking)是基于结构药物设计的核心模拟手段,依据受体与配体作用时的几何匹配和能量匹配过程,模拟受体-配体相互作用,预测两者间最佳的结合模式和结合亲和力。采用分子对接模拟技术,科研人员可以进行基于结构的药物虚拟筛选,药物分子的结构改造,配体和受体相互作用的机理研究等工作,从而大大提高实验效率。在Discovery Studio这一分子模拟的综合平台中,分子对接程序包含Libdock、CDOCKER、Flexible Docking,这三种对接算法各有优势,能够满足广大科研工作者的多种应用需求,为其提供配体受体间相互识别的“利器”。
鬼臼毒素通过减少DNA损伤和减少PI3K抑制作用可作为抗肿瘤候选药物
Ref:Journal of Medicinal Chemistry. Published: February 21, 2020,IF:6.205
链接:https://dx.doi.org/10.1021/acs.jmedchem.9b01354
替尼泊苷(4,6-O-亚苯基-β-D-吡喃葡萄糖苷-4-去甲基表鬼臼毒素)是4-脱甲基表鬼臼毒素(DMEP)的半合成糖缀合物,在1992年被美国FDA批准用于临床癌症化疗。在过去的几十年中,几乎所有研究都集中在发现比替尼泊苷更有效的新型先导化合物上。实际上,替尼泊苷或其他DMEP衍生物的化疗瓶颈是严重的DNA毒性。毒性机理表明,替尼泊苷可以与拓扑异构酶II (Topo II)紧密结合,并阻断转运蛋白生成的DNA双链断裂(DSBs)。研究表明多种给药方式也并未降低替尼泊苷的毒性。为了降低毒性,分子结构修饰是最基本、最有效的方法。结合之前的工作发现通过修饰DMEP的分子结构,发现了具有毫摩尔毒性的有效抗肿瘤前导化合物。毒性研究表明,5-氟苯并噻唑和5-氟苯并恶唑可通过特异性缓解和修复DNA双链断裂来提高DMEP的抗肿瘤活性,而对正常HL-7702细胞没有DNA毒性。替尼泊苷是Topo II典型的药物,但是具有较高的细胞毒性。为了发现安全的第二代药物,本研究旨在设计和合成2’或3’酰胺化或酯化-苯并杂环取代的替尼泊苷衍生物,以发现具有弱DNA毒性的新型先导化合物。
首先通过分子对接将替尼泊苷对接到DNA拓扑异构酶II中,对接结果表明替尼泊苷稳定地结合在Topo II-DNA复合物中,并不可逆地阻止了另一条DNA链与DNA骨架的重新连接。因此,替尼泊苷具有良好的抗肿瘤活性,但也引起了过度的毒性。作者假设替尼泊苷的毒性降低可通过改变替尼泊苷与Topo II- DNA复合物的结合模式,通过阻断Topo II的结合位点作为非嵌入的Topo II催化抑制剂。结合替尼泊苷结构活性关系(SAR),作者猜测对糖苷部分的2’或3’位修饰会适度降低替尼泊苷的毒性。根据拓扑异构酶II的切割DNA结合结构域,选择5-氟苯并噻唑和5-苯并恶唑作为弱氢键受体以修饰替尼泊苷的分子结构,以适度降低切割DNA结合亲和力,共设计20种替尼泊苷衍生物,通过生物实验验证化合物15和16与替尼泊苷相比具有较低的细胞毒性。总之,通过分子对接可以为先导化合物的修饰和改造提供一定的思路。
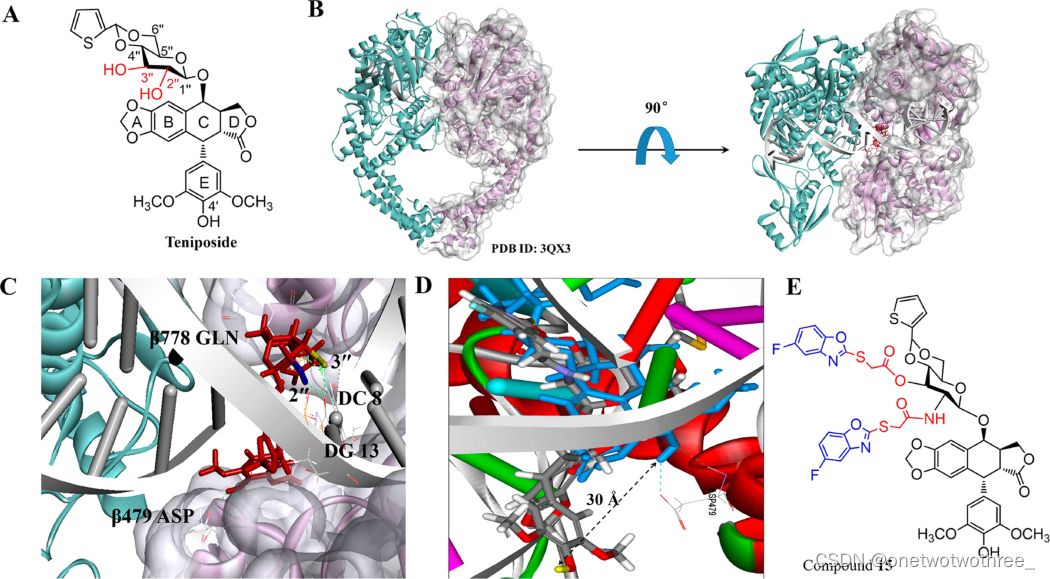
图1 DNA双链断裂结合位点与化合物的结合模式图
(A) 替尼泊苷的分子结构;(B) 化合物15与拓扑异构酶II-DNA相互作用分析图;(C) 替尼泊苷与拓扑异构酶II的DNA断裂域相互作用图;(D)替尼泊苷和化合物15与DNA断裂域的相互作用的比较;(E) 化合物15的分子结构。
MaXFlow分子模拟与人工智能平台
MaXFlow生物医药智能创新平台,由创腾科技自主研发,旨为不同领域的一线创新科技工作者提供一个合作共享的B-S架构平台。以“数据自由,模型自由”为理念,在结构模型与预测模型进行融合的基础上,实现模拟与AI需求的合并,为研发赋能。
- 填补数据产生保存与数据使用赋能断层
- 打通空间结构模型与数据预测模型壁垒
- 合并经典模拟计算与新兴AI预测需求
- 降低背景知识储备与复杂软件使用门槛
通过便捷的网页端操作,可实现大、小分子模型的构建与优化,动力学模拟,分子对接,分子间相互作用展示。小分子药物方面,通过分子性质计算以及多种机器学习与深度学习的方法,在工作流中帮助用户实现数据的挖掘以及相关构效关系的搭建,同时可以通过一键部署的方式实现药代动力学及不同目的的AI预测与共享。对于大分子,基于流行AI模型的运用,更加准确的实现大分子间相互作用预测。多样的APPs为大分子药物研发提供可靠保障。
本文来自互联网用户投稿,文章观点仅代表作者本人,不代表本站立场,不承担相关法律责任。如若转载,请注明出处。 如若内容造成侵权/违法违规/事实不符,请点击【内容举报】进行投诉反馈!